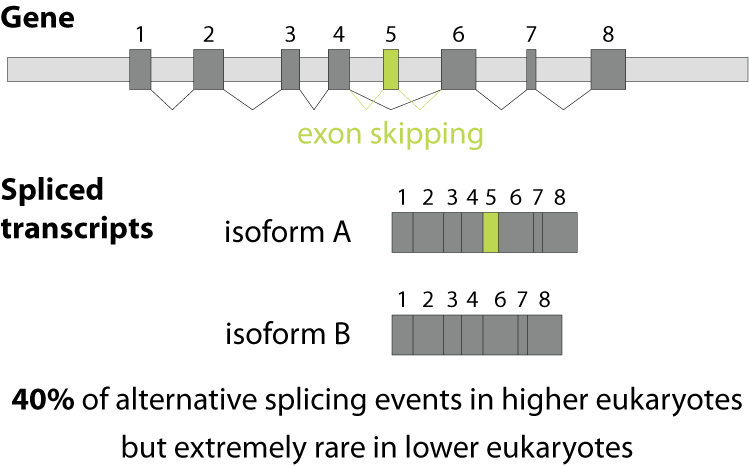
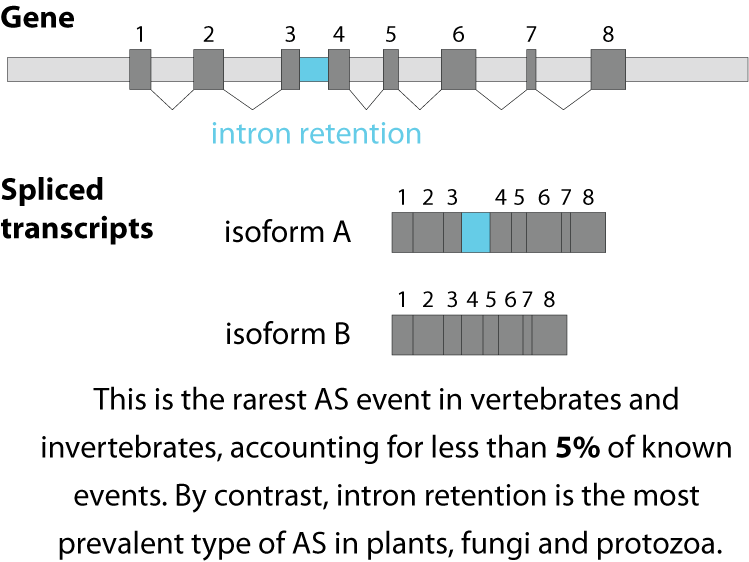
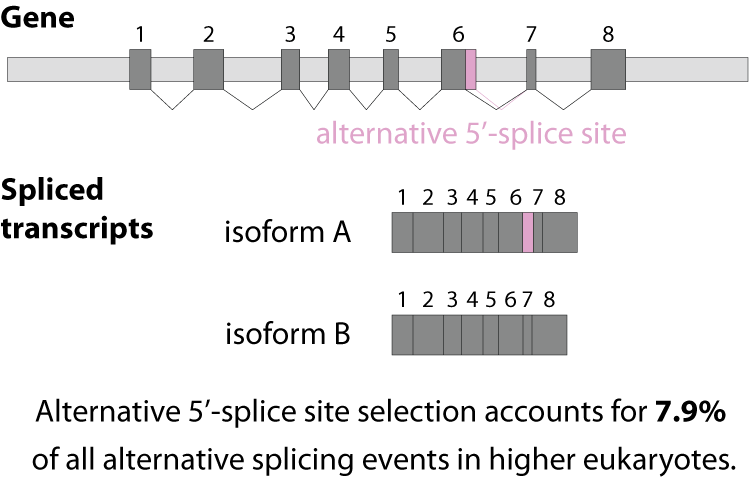
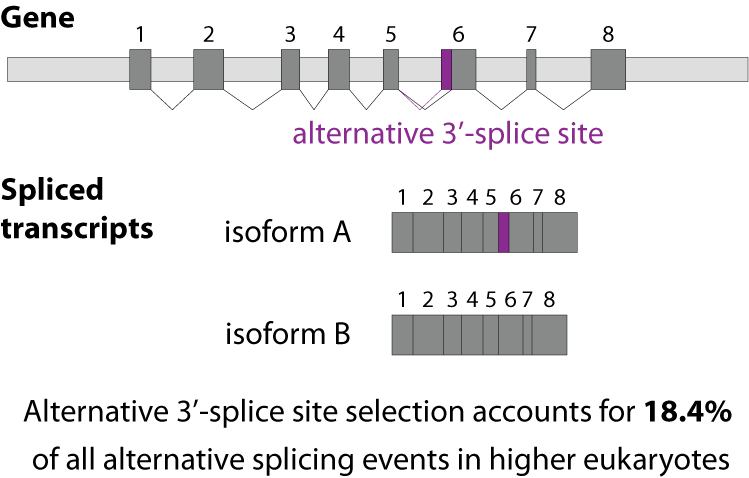
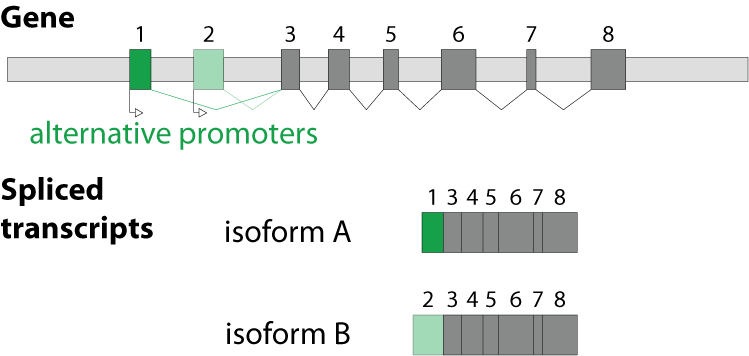
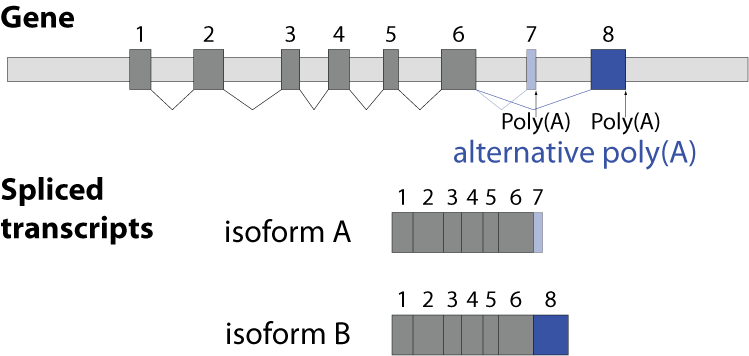
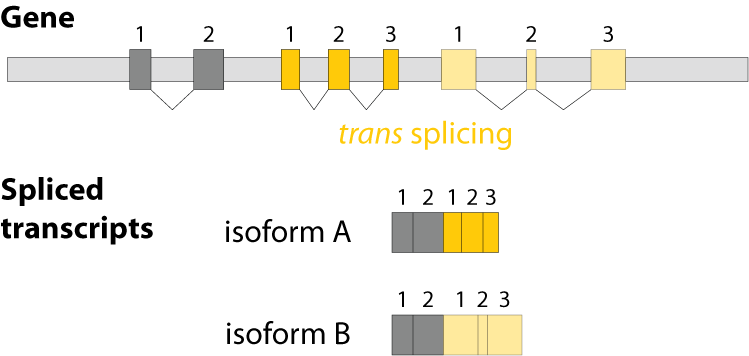
Alternative splicing is an important process in higher eukaryotes that allows obtaining several transcripts from one gene. Presumably every single human gene containing more than one exon is transcribed and processed to yield multiple mRNAs. Mainly, five different types of alternative splicing affect the resulting translated protein product: The first type is exon skipping, in which an exon, also called cassette exon, is spliced out of the transcript together with its flanking introns. The second and third types are the alternative splicing of the 3’ splice site and 5’ splice site, respectively. Here, two or more splice sites are recognized at one end of the exon. The fourth type is intron retention in which part of an exon is either spliced (like a regular intron) or retained in the mature mRNA transcript.
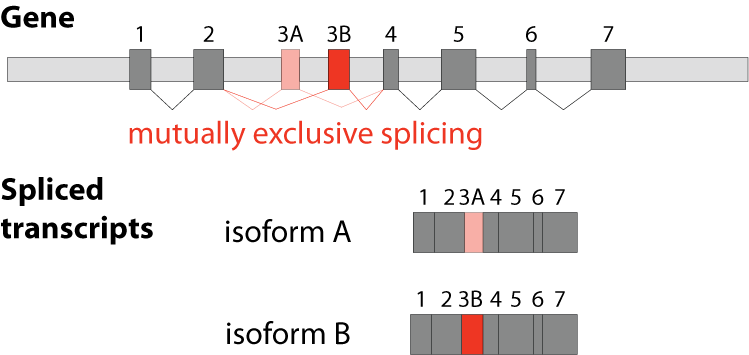
The fifths type is called mutually exclusive splicing and is used for clusters of internal exons that are spliced in a mutually exclusive manner. The following constrains apply to mutually exclusive exons (MXEs): A) Mutually exclusive exons must have about the same length (allowing some length difference for e.g. parts encoding loop regions). B) They must have conserved splice site patterns (e.g. a U1/U2 intron splice site cannot be combined with a U11/U12 splice site) and the reading frame of the exons must be conserved. C) MXEs must show sequence similarity.
Although MXEs within a cluster are relatively similar, they cannot substitute each other if one is damaged. MXEs have been described in many crucial and essential human genes such as in the α-subunits of six of the ten voltage-gated sodium channels (SCN genes), in each of the glutamate receptor subunits 1-4 (GluR1-4) where the MXEs are called flip and flop, and in SNAP-25 as part of the neuroexocytosis machinery. Mutations in MXEs have been shown to cause diseases such as Timothy syndrome (missense mutation in the CACNA1C gene), cardiomyopathy (defect of the mitochondrial phosphate carrier SLC25A3) or cancer (mutations in e.g. the pyruvate kinase PKM and the zinc transporter SLC39A14).
Recently, we predicted 1722 completely novel exons in previously intronic regions in human (Hatje et al., 2017, Mol Sys Biol). By mapping 15 billion RNA-Seq reads, selecting 515 samples comprising 31 tissues and organs, 12 cell lines and 7 developmental stages, we could show that all exons are covered by at least one read. Applying strict criteria requiring reads bridging the respective other MXE and the absence of reads joining MXEs, 1399 of the 6541 MXE candidates comprise high-confidence MXEs. This number is about 10-fold higher than the previously known MXEs in human. One aspect of special importance is the role of MXEs in human disease. Disease linkage is often analysed using exome arrays or sequencing, linking known exons and genes to putative disease loci containing SNPs or other structural variants. Consequentially, exome analysis is blind to intergentic, intronic, and unannotated exonic mutations, as is the case for the newly predicted, previously uncharacterized exons. This directly implies that previously ‘intronic’ disease-associated SNPs might actually be ‘exonic’, and might thereby change crucial residues in the respective protein product. To assess the relevance of MXEs in human disease, we overlayed the set of MXE candidates with the ClinVar database. This resulted in 35 MXEs (eight ‘novel exons’) with 82 pathogenic SNPs, which reflects the constraints of exome sequencing mentioned above. Disease-associated MXEs show tight developmental and tissue-specific expression (average Gini-index of 0.65) with prominent selective expression in heart and brain, and cancer cell lines.
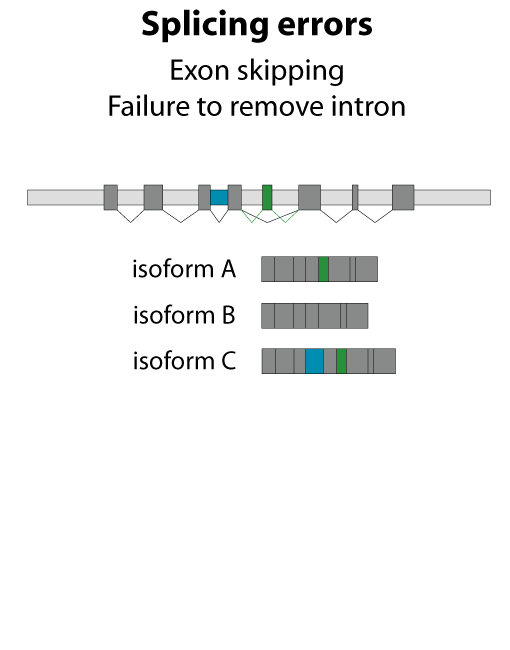
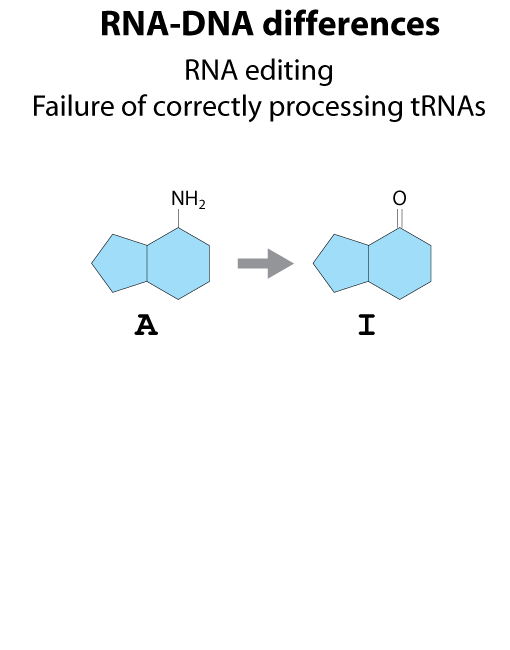
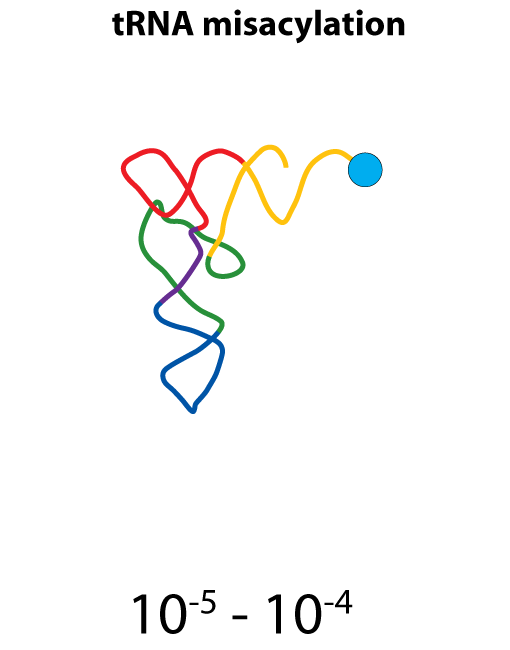
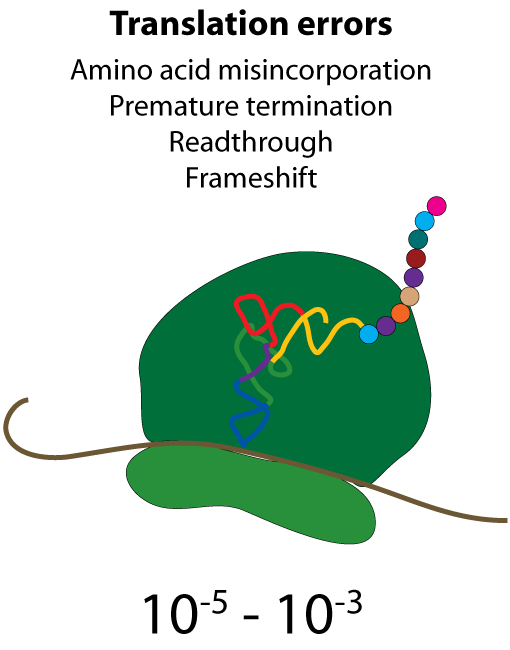
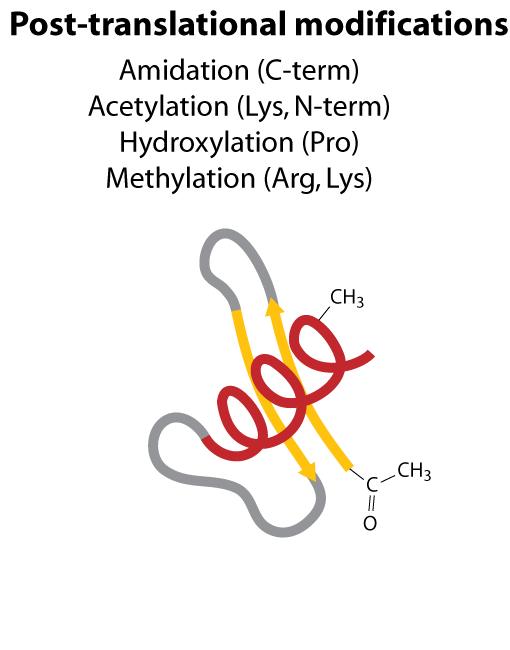
All steps in the process of synthesizing a functional protein from genetic information are error prone. Disruption in the conversion of coding sequence into a functional protein can happen during transcription (nucleotide misincorporation, polymerase slippage), splicing (exon skipping, failure to remove intron), translation (tRNA misacylation, amino acid misincorporation, frameshift, readthrough), folding (kinetic trapping, spontaneous unfolding) and post-translational modifications.

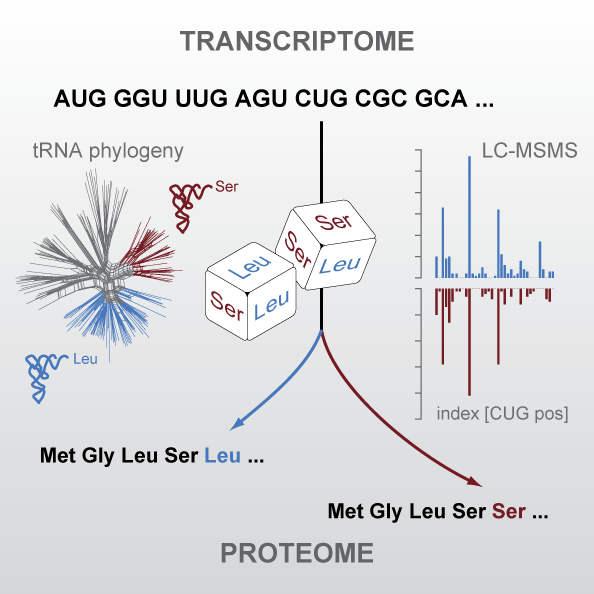
The universal genetic code defines the translation of nucleotide triplets, called codons, into amino acids. In many Saccharomycetes a unique alteration of this code affects the translation of the CUG codon, which is normally translated as leucine. Most of the species encoding CUG alternatively as serine belong to the Candida genus and were grouped into a so-called CTG clade. By generating proteomics data and using tRNA sequence comparisons, we showed that another yeast, Pachysolen tannophilus, translates CUG codons as alanine. Despite all these alterations to the ‘‘universal’’ genetic code demonstrating it not to be universal one regularity remained, namely that for a given sense codon there is a unique translation. However, examining CUG usage in yeasts that have transferred CUG away from leucine, we reported the first example of dual coding: Ascoidea asiatica stochastically encodes CUG as both serine and leucine in approximately equal proportions. This is deleterious, as evidenced by CUG codons being rare, never at conserved serine or leucine residues, and predominantly in lowly expressed genes. Related yeasts solvee the problem by loss of function of one of the two tRNAs. This dual coding is consistent with the tRNA-loss-driven codon reassignment hypothesis, and provides a unique example of a proteome that cannot be deterministically predicted.