Scipio / WebScipio - Eukaryotic gene identification
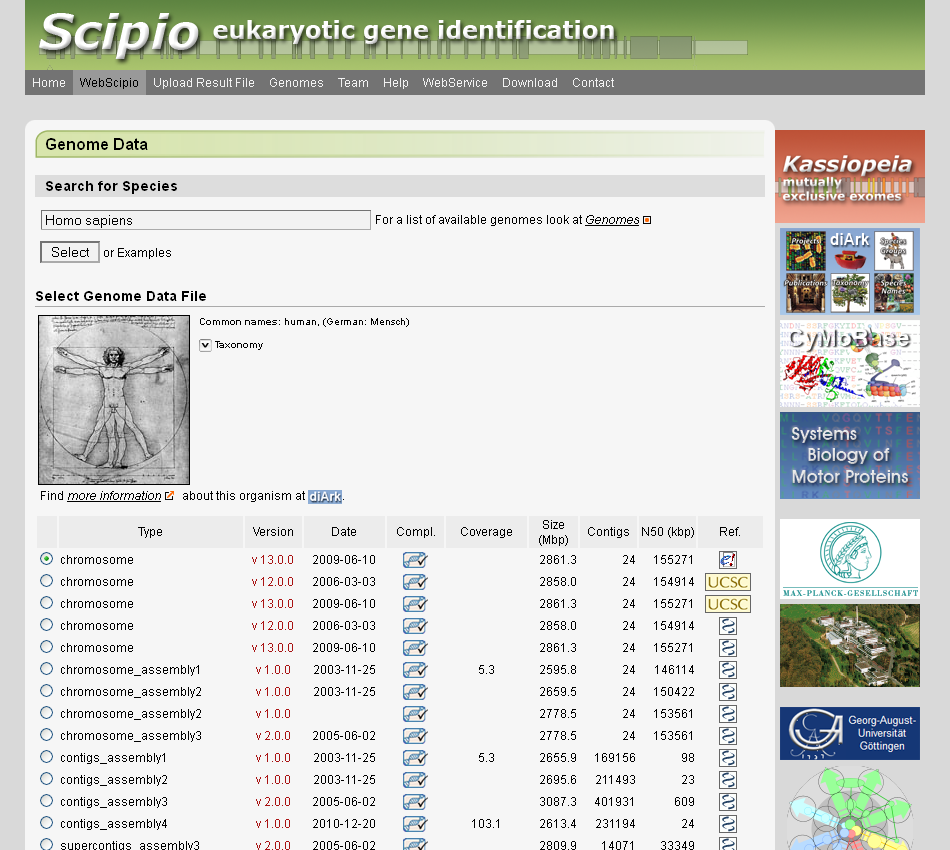
Accurate exon–intron structures are essential prerequisites in genomics and proteomics, and for many protein family and single gene studies. We originally developed Scipio and the corresponding web service WebScipio for the reconstruction of gene structures based on protein sequences and available genome assemblies. WebScipio also allows predicting mutually exclusive spliced exons and tandemly arrayed gene duplicates. The obtained gene structures are illustrated in graphical schemes and can be analysed down to the nucleotide level. The set of eukaryotic genomes available at the WebScipio server is updated regularly. The current version of the web server provides access to more than 13,100 genome assembly files of more than 6,300 sequenced eukaryotic species. WebScipio is freely available at webscipio.org.
Kassiopeia - Analysing mutually exclusive exomes
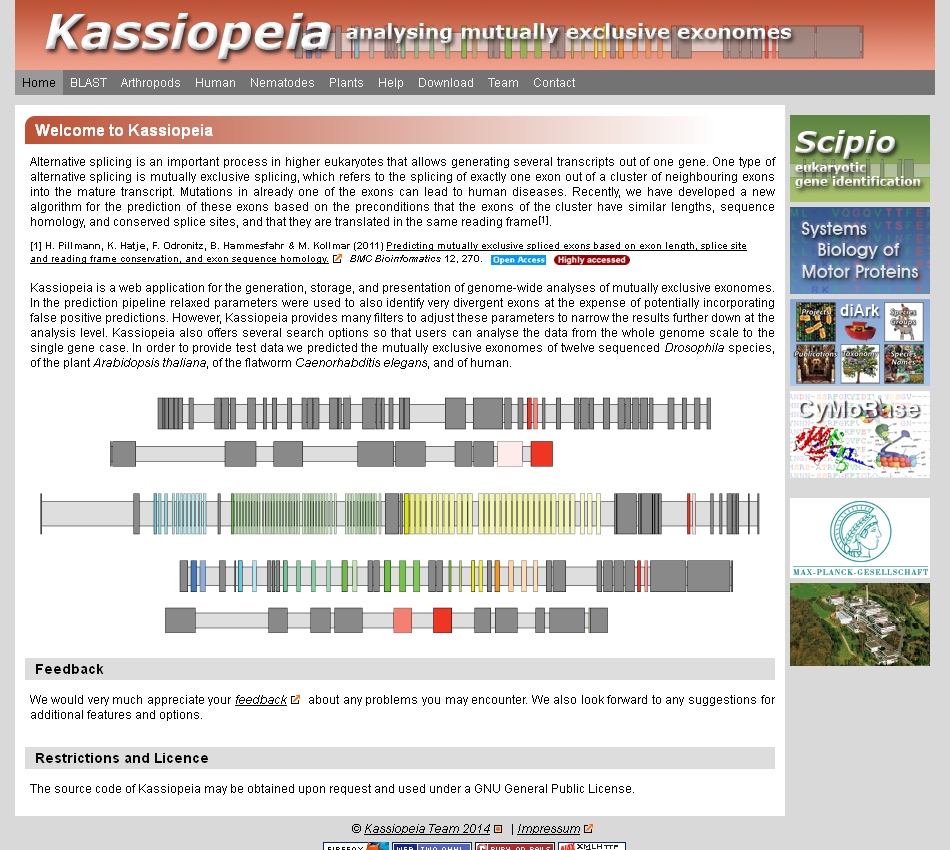
Kassiopeia is a database and web application for the generation, storage, and presentation of genome-wide analyses of mutually exclusive exomes. Currently, Kassiopeia provides access to the mutually exclusive exomes of twelve Drosophila species, the thale cress Arabidopsis thaliana, the flatworm Caenorhabditis elegans, and human. Mutually exclusive spliced exons (MXEs) were predicted based on gene reconstructions from Scipio. The user can search Kassiopeia using BLAST or browse the genes of each species, optionally adjusting the parameters used for the prediction to reveal more divergent or only very similar exon candidates. For each gene Kassiopeia provides a comprehensive gene structure scheme, the sequences and predicted secondary structures of the MXEs, and, if available, further evidence for MXE candidates from cDNA/EST data, predictions of MXEs in homologous genes of closely related species, and RNA secondary structure predictions. Kassiopeia can be accessed at kassiopeia.motorprotein.de.
diArk - A resource for eukaryotic genome research
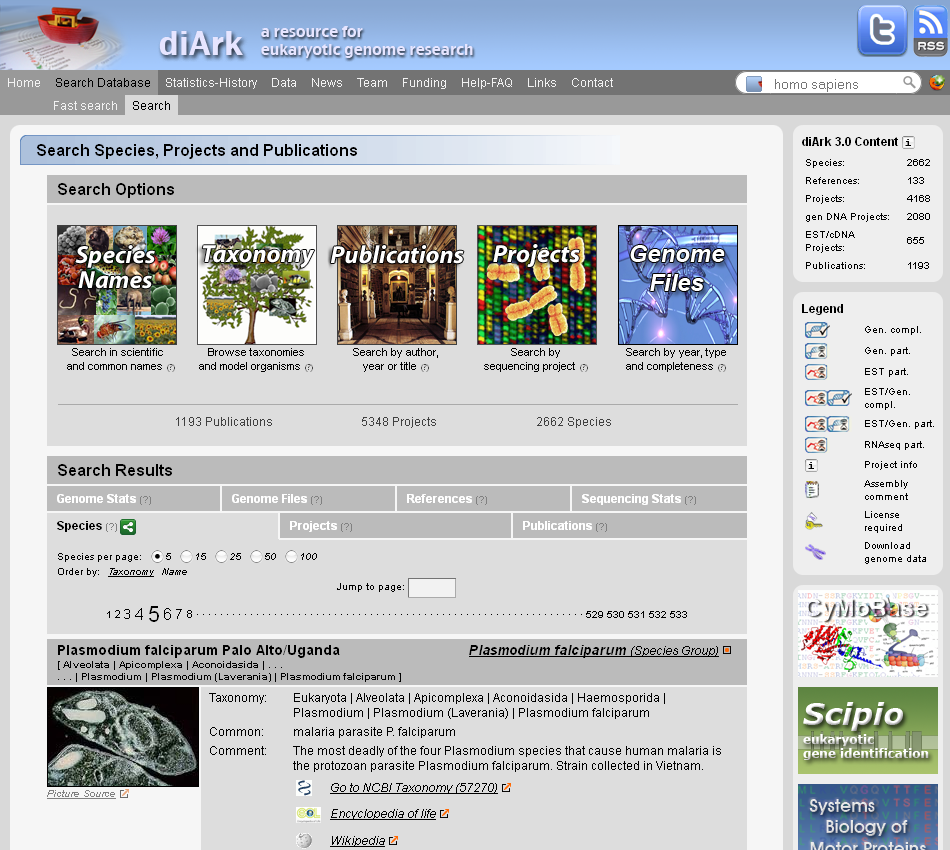
Eukaryotic genomes are the basis for understanding the complexity of life from populations to the molecular level. Recent technological innovations have revolutionized the speed of data generation enabling the sequencing of eukaryotic genomes and transcriptomes within days. The database diArk has been developed with the aim to provide access to all available assembled genomes and transcriptomes. In September 2018, diArk contains about 6,500 eukaryotes with 13,000 genome and transcriptome assemblies, of which about 22% are not available via NCBI/ENA/DDBJ. Several indicators for the quality of the assemblies are provided to facilitate their comparison for selecting the most appropriate dataset for further studies. diArk has a user-friendly web interface with extensive options for filtering and browsing the sequenced eukaryotes. diArk is freely available at diark.org.
CyMoBase - A database for cytoskeletal and motor proteins
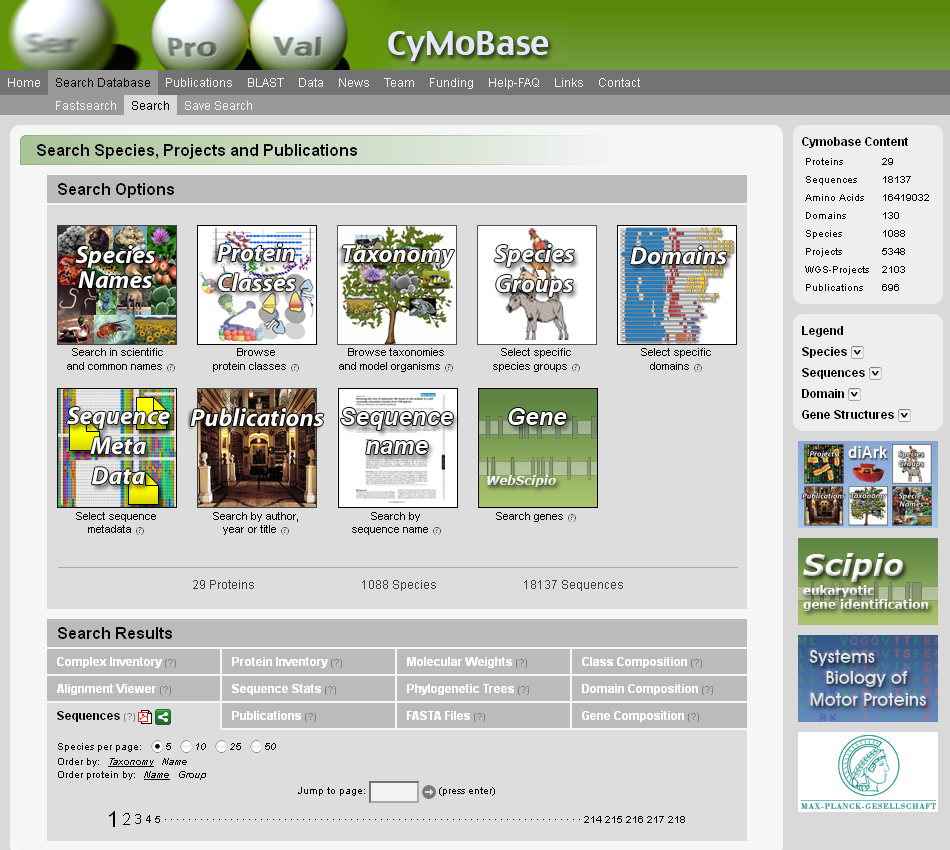
Annotation of protein sequences of eukaryotic organisms is crucial for the understanding of their function in the cell. Manual annotation is still by far the most accurate way to correctly predict genes. The classification of protein sequences, their phylogenetic relation and the assignment of function involves information from various sources. This often leads to a collection of heterogeneous data, which is hard to track. Cytoskeletal and motor proteins consist of large and diverse superfamilies comprising up to several dozen members per organism. Since genome sequence data is rapidly accumulating it is very important to have a reference database for the nomenclature and phylogenetic relation of the proteins that allows the most accurate assignment of biological function possible. CyMoBase is a protein sequence-centric web application to store, organize, interrelate, and present heterogeneous data that is generated during manual genome annotation and comparative genomics. It offers many analysis tools like extensive statistics or a BLAST service. CyMoBase is freely available at cymobase.org.
Bagheera - Predicting CUG codon translation in yeasts

The universal genetic code defines the translation of nucleotide triplets, called codons, into amino acids. In many Saccharomycetes a unique alteration of this code affects the translation of the CUG codon, which is usually translated as leucine. Most of the species encoding CUG alternatively as serine belong to the Candida genus and were grouped into a so-called CTG clade. However, the “Candida genus” is not a monophyletic group and several Candida species are known to use the standard CUG translation. By performing a phylogenomics analysis of 26 motor and cytoskeletal proteins from 60 sequenced yeast species and by investigating the CUG codon positions with respect to sequence conservation at the respective alignment positions, we were able to unambiguously assign the standard code or Alternative Yeast Codon Usage (AYCU). These data are the basis for the Bagheera webserver, which can be used to detect the most probable translation scheme of a given yeast species. Bagheera can freely be accessed at bagheera.motorprotein.de.
GenePainter - Aligning gene structures
All sequenced eukaryotic genomes have been shown to possess at least a few introns including unicellular organisms, which were previously suspected to be intron-less. Therefore, gene splicing must have been present at least in the last common ancestor of the eukaryotes. To explain the evolution of introns, basically two mutually exclusive concepts have been developed. The introns-early hypothesis says that already the very first protein-coding genes contained introns while the introns-late concept asserts that eukaryotic genes gained introns only after the emergence of the eukaryotic lineage. A very important aspect in this respect is the conservation of intron positions within homologous genes of different taxa. GenePainter is a standalone application for mapping gene structure information onto protein multiple sequence alignments. Based on the multiple sequence alignments the gene structures are aligned down to single nucleotides. GenePainter accounts for variable lengths in exons and introns, respects split codons at intron junctions and is able to handle sequencing and assembly errors, which are possible reasons for frame-shifts in exons and gaps in genome assemblies. Conserved intron positions can also be mapped to user-provided protein structures. GenePainter can also be accessed via a web interface. GenePainter is freely available at genepainter.motorprotein.de.
Waggawagga - Coiled-coil and single-alpha-helix domain prediction
Coiled-coil predictions are characterized by contiguous heptad repeats, which can be depicted in the form of a net-diagram. From this representation a score has been developed, which enables the discrimination between coiled-coil-regions and single α-helices. The software was implemented as a web-application and a command-line tool. The user can run applications for the sole prediction of coiled-coils and applications for the prediction of the oligomerisation states. In the web interface, the predicted coiled-coil sequences are visualized as helical wheel-diagram of parallel or anti-parallel homodimers, or parallel homotrimers, and as heptad-net-diagram. In addition, the SAH-score is calculated for each prediction. Considered together, these information provide an indication for the correct prediction of the structural motives. Waggawagga is freely available at waggawagga.motorprotein.de.
Peakr - simulating solid-state NMR spectra of proteins
When analyzing solid-state nuclear magnetic resonance (NMR) spectra of proteins, assignment of resonances to nuclei and derivation of restraints for 3D structure calculations are challenging and time-consuming processes. Simulated spectra that have been calculated based on, for example, chemical shift predictions and structural models can be of considerable help. Existing solutions are typically limited in the type of experiment they can consider and difficult to adapt to different settings. Here, we present Peakr, a software to simulate solid-state NMR spectra of proteins. It can generate simulated spectra based on numerous common types of internuclear correlations relevant for assignment and structure elucidation, can compare simulated and experimental spectra and produces lists and visualizations useful for analyzing measured spectra. Compared with other solutions, it is fast, versatile and user friendly. Peakr is freely available at peakr.org.
ShereKhan - calculating molecular exchange rates
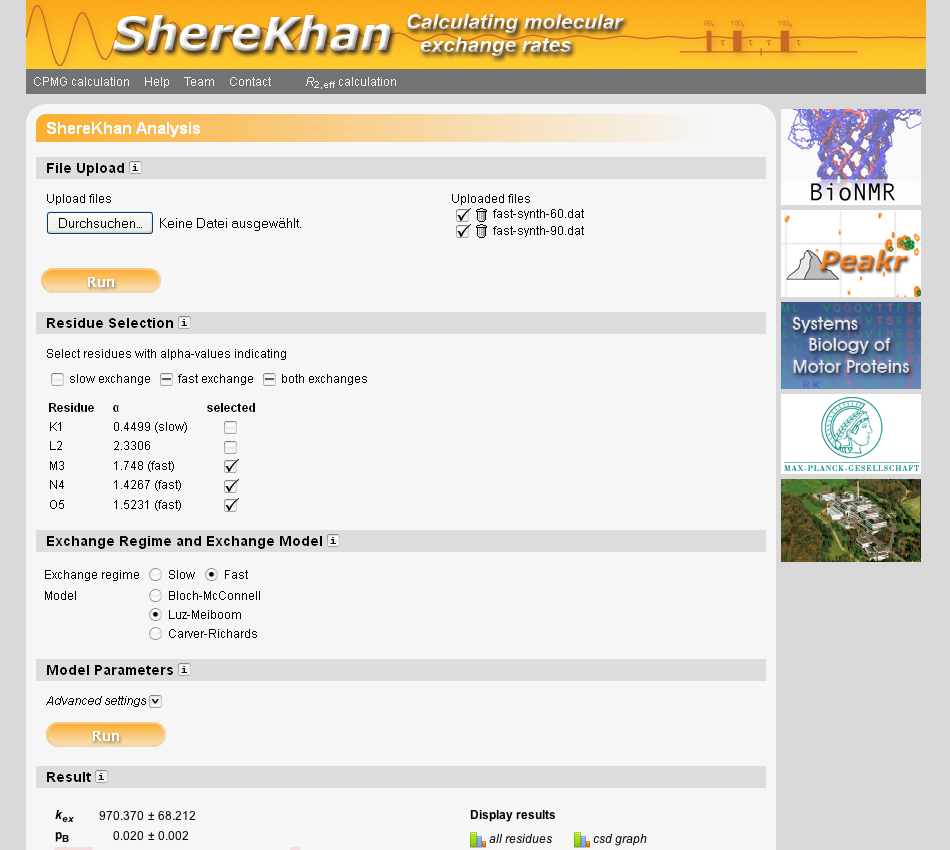
Dynamics governing the function of biomolecule are usually described as exchange processes and can be monitored at atomic resolution with nuclear magnetic resonance (NMR) relaxation dispersion data. Here, we present a new tool for the analysis of CPMG relaxation dispersion profiles (ShereKhan). The web interface to ShereKhan provides a user-friendly environment for the analysis. Sherekhan can freely be accessed at sherekhan.bionmr.org.



